Our Reseach
Research contributions from the Wilson lab have been pivotal for many of the pioneering studies on immune recognition of microbial pathogens, antigen presentation and structure-based design of vaccine and therapies. We have structurally characterized many key antigen recognition receptors in innate and adaptive immunity, including T cell receptors, MHC class I and II, over 350 antibodies and antibody complexes, CD1, TLRs, VLRs, NODs, etc. Our research in structural virology has focused on structural and functional characterization of the challenging glycoprotein antigens from influenza virus, HIV-1, and HCV, and the circumsporozoite protein from P. falciparum. Our recent work has been on identifying the sites of vulnerability on these microbial pathogens that are targeted by broadly neutralizing and protective antibodies, why these antibodies are so effective, and how we can use this information for structure-based design of vaccines and therapies. Dr Wilson also directed the Joint Center for Structural Genomics (JCSG) for 16 years (2000-2016) that pioneered high-throughput methods for protein expression, crystallization and structure determination and deposited over 1600 novel structures into the Protein Data Bank (PDB), which greatly expanded the universe of known protein structures. Our lab operates a HT protein crystallization facility that was formerly designed and operated by JCSG and houses the latest equipment and tools for biophysical characterization of proteins, including a fully automated protein crystallization robots CrystalMation system.
 Innate and adaptive immune receptors. Innate and adaptive immunity provides defense against pathogen- or danger-associated molecular receptors. We determined the first structure of a T-cell receptor in complex with pMHC which revealed how peptide antigens are recognized in the context of the MHC molecule (Fremont et al. 1992; Garcia et al. 1996). We have also studied other key molecules in cellular immunity, such as non-classical or MHC homologues, including how CD1 binds to lipid, glycolipid, and lipopeptide antigens from the cell walls of microbial pathogens and the NK family of receptors recognizes classical as well as distant MHC homologues. Other pattern recognition receptors investigated include peptidoglycan recognition protein (PGRP) and TREM-1. TLRs have revealed how unique pathogenassociated molecules are recognized by the immune system (Choe et al. 2005; Yoon et al. 2012) and VLRs provide an alternate solution for recognition of antigens (Han et al. 2008). Toll-like receptors (TLRs), VLRs and NOD-like receptors (NLRs) share a common structural motif that consists of multiple leucine-rich repeats (LRRs) with some variability in amino-acid residues and length of each repeat. Structures of LRRs in TLRs, VLRs, and NLRs by themselves or complexed with their ligands or signaling partners are advancing our understanding of receptor activation and regulatory mechanisms in innate immunity and the development of agonists and antagonists as adjuvants, or as therapeutics in compromised immunity or in hyper-inflammatory diseases.
Innate and adaptive immune receptors. Innate and adaptive immunity provides defense against pathogen- or danger-associated molecular receptors. We determined the first structure of a T-cell receptor in complex with pMHC which revealed how peptide antigens are recognized in the context of the MHC molecule (Fremont et al. 1992; Garcia et al. 1996). We have also studied other key molecules in cellular immunity, such as non-classical or MHC homologues, including how CD1 binds to lipid, glycolipid, and lipopeptide antigens from the cell walls of microbial pathogens and the NK family of receptors recognizes classical as well as distant MHC homologues. Other pattern recognition receptors investigated include peptidoglycan recognition protein (PGRP) and TREM-1. TLRs have revealed how unique pathogenassociated molecules are recognized by the immune system (Choe et al. 2005; Yoon et al. 2012) and VLRs provide an alternate solution for recognition of antigens (Han et al. 2008). Toll-like receptors (TLRs), VLRs and NOD-like receptors (NLRs) share a common structural motif that consists of multiple leucine-rich repeats (LRRs) with some variability in amino-acid residues and length of each repeat. Structures of LRRs in TLRs, VLRs, and NLRs by themselves or complexed with their ligands or signaling partners are advancing our understanding of receptor activation and regulatory mechanisms in innate immunity and the development of agonists and antagonists as adjuvants, or as therapeutics in compromised immunity or in hyper-inflammatory diseases.
Select Publications
- Structural basis of TLR5-flagellin recognition and signaling. Science. 2012; 335:859-64. PMCID: PMC3406927.
- Antigen recognition by variable lymphocyte receptors. Science 2008; 32:1834-7. PMCID: PMC2581502.
- Crystal structure of human toll-like receptor 3 (TLR3) ectodomain. Science 2005; 309:581-5. PMID: 15961631.
- An alphabeta T cell receptor structure at 2.5 A and its orientation in the TCR-MHC complex. Science 1996; 274:209-19. PubMed PMID: 8824178.
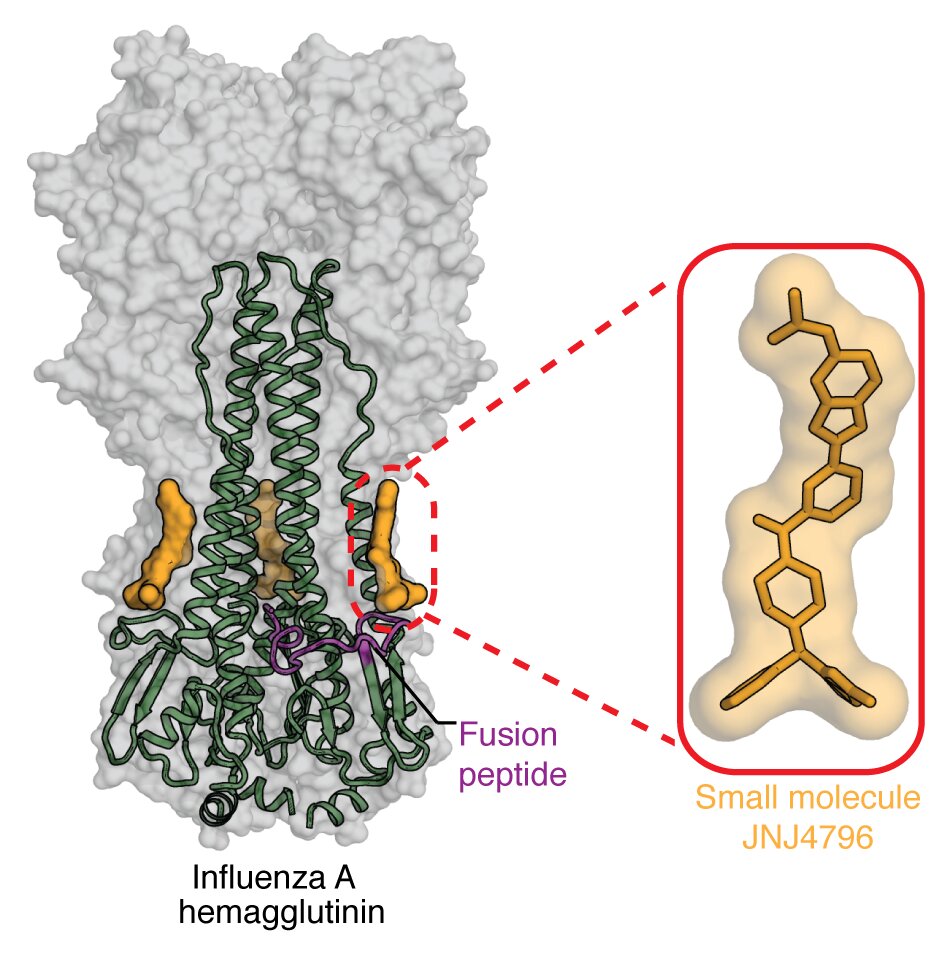 Influenza antigen structures, sites of vulnerability, universal vaccines and therapy. The structure of the hemagglutinin (HA) major surface antigen of influenza virus in 1981 was a breakthrough in the virology field that enabled understanding of its function with respect to its receptor binding and fusion activities and how influenza can escape from the immune system (Wilson et al., Nature 1981;289:366-73). This first viral glycoprotein structure included 25% carbohydrate by weight. Many other HA structures from my lab include influenza A 1918 H1, H2, H5, H6, H7, H9, H10, H15 and flu B HAs that illustrate the diversity in influenza subtypes, as well as various N2, N7, N10, N11 NA structures. We have focused recently on how influenza virus is recognized by human broadly neutralizing antibodies (bnAbs). For example, bnAb CR6261 broadly targets influenza virus through binding a conserved epitope in the HA stem required for viral fusion. This work gave renewed hope for a universal flu vaccine (Ekiert et al., Science 2009;324:246-51) and spurred searches for other bnAbs, including CR9114 that binds to influenza A and B viruses (Dreyfus et al., Science 2012;337:1343-8). Recently, bnAbs to influenza neuraminidase (Stadlbauer et al., Science 2019 366: 409-504) have renewed interest in NA as a vaccine target. Through determination of many bnAb structures, we have thereby elucidated common features and motifs that impart breadth and potency and provided valuable insights for design of universal vaccines (1), universal multidomain antibody immunotherapy (2), and small molecule therapeutics (3, 4).
Influenza antigen structures, sites of vulnerability, universal vaccines and therapy. The structure of the hemagglutinin (HA) major surface antigen of influenza virus in 1981 was a breakthrough in the virology field that enabled understanding of its function with respect to its receptor binding and fusion activities and how influenza can escape from the immune system (Wilson et al., Nature 1981;289:366-73). This first viral glycoprotein structure included 25% carbohydrate by weight. Many other HA structures from my lab include influenza A 1918 H1, H2, H5, H6, H7, H9, H10, H15 and flu B HAs that illustrate the diversity in influenza subtypes, as well as various N2, N7, N10, N11 NA structures. We have focused recently on how influenza virus is recognized by human broadly neutralizing antibodies (bnAbs). For example, bnAb CR6261 broadly targets influenza virus through binding a conserved epitope in the HA stem required for viral fusion. This work gave renewed hope for a universal flu vaccine (Ekiert et al., Science 2009;324:246-51) and spurred searches for other bnAbs, including CR9114 that binds to influenza A and B viruses (Dreyfus et al., Science 2012;337:1343-8). Recently, bnAbs to influenza neuraminidase (Stadlbauer et al., Science 2019 366: 409-504) have renewed interest in NA as a vaccine target. Through determination of many bnAb structures, we have thereby elucidated common features and motifs that impart breadth and potency and provided valuable insights for design of universal vaccines (1), universal multidomain antibody immunotherapy (2), and small molecule therapeutics (3, 4).
Select Publications
- A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 2015; 349:1301-6. PMID: 26303961.
- Universal protection against influenza infection by a multidomain antibody to influenza hemagglutinin. Science 2018;362:598-602. PMID: PMC6241527.
- Potent peptidic fusion inhibitors of influenza virus. Science 2017;358:496-502. PMCID: PMC5659926.
- A small-molecule fusion inhibitor of influenza virus is orally active in mice. Science. 2019;363:eaar6221. PMCID: PMC6457909.
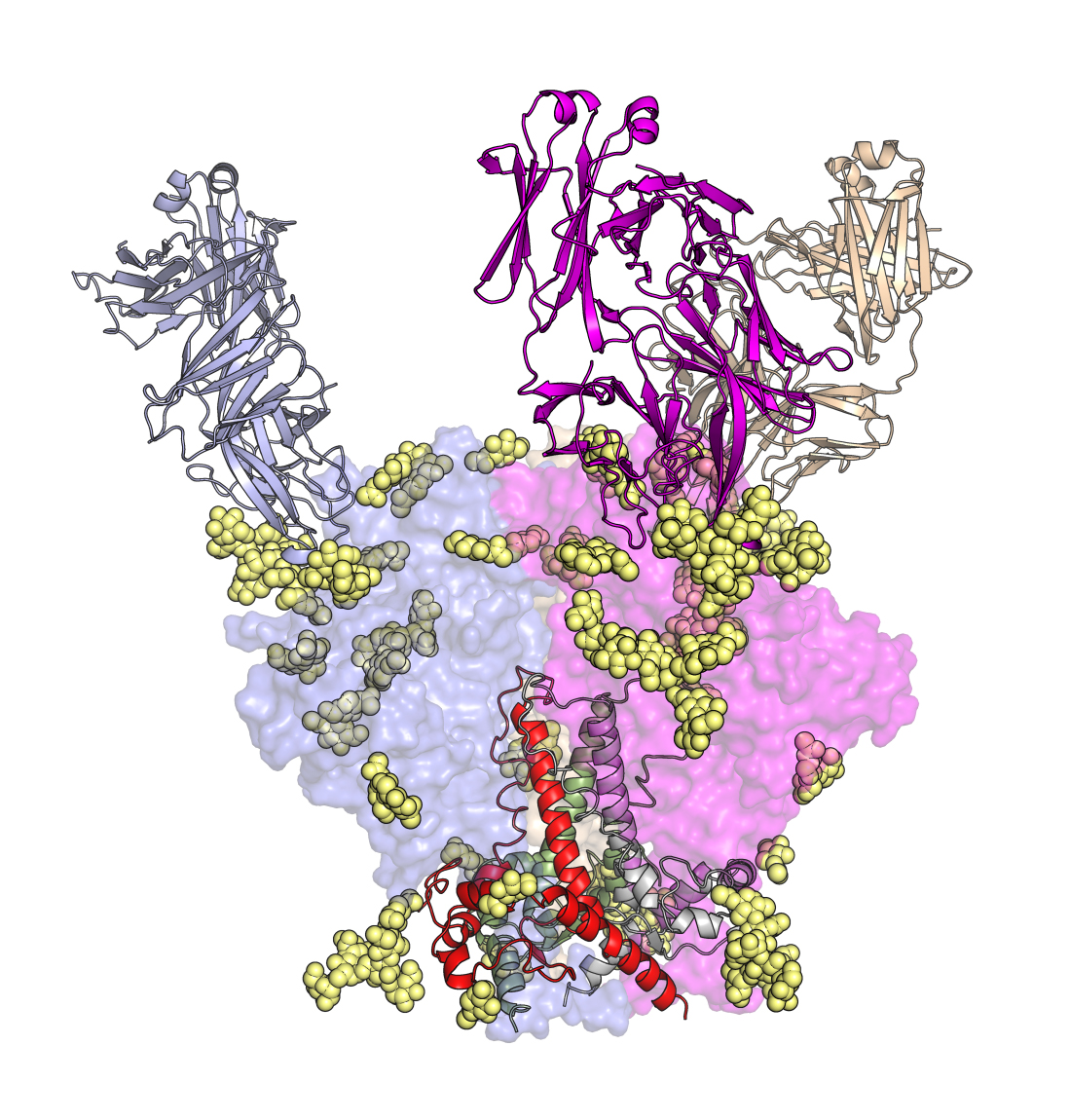 HIV-1 antigen structure, sites of vulnerability and structure-assisted vaccine design. The quest for a structure of the HIV-1 Envelope glycoprotein (Env) has been a holy grail in structural biology and virology and was cited as one of the 5 key structures still to be determined by the Nature journals. The challenge was to obtain sufficient soluble, native-like material for structural studies. Over a 12 year collaboration with John Moore’s group at Weill Medical College, we were able to determine the crystal structure of a soluble cleaved Env trimer (SOSIP) (1) that enabled a view of the interaction between the component gp41 and gp120 subunits, assembly of the three gp140s that compose the trimer, and its interaction with bnAbs. This work with a companion cryo-EM structure of the same trimer (Lyumkis et al., Science 342:1484-90, 2013) defined key features of the Env trimer and that it was a faithful mimic of Env on the virus, and both papers were cited as runner up papers of the year in Science in 2014. Our current focus is how HIV-1 Env is recognized by bnAbs to define sites of vulnerability for structure-assisted vaccine design (e.g. Pejchal et al., Science 334:1097-1103, 2011 and 2). These bnAbs almost all also target glycans on the Env surface (3). The Env structure has propelled its use as a candidate immunogen and many groups are using this soluble BG505 SOSIP Env trimer construct for vaccine design, such as NFL here, and for testing in animal models. We generated responses against the SOSIP trimer as a candidate immunogen in small animal models and, for the first time, a tier-2 neutralizing response against the homologous virus (Sanders et al., Science 349:aac4223, 2015), thus showing progress towards design of an HIV-1 vaccine (reviewed 4). Indeed, this BG505 SOSIP Env trimer has entered a Phase 1 human clinical trial (March 2019).
HIV-1 antigen structure, sites of vulnerability and structure-assisted vaccine design. The quest for a structure of the HIV-1 Envelope glycoprotein (Env) has been a holy grail in structural biology and virology and was cited as one of the 5 key structures still to be determined by the Nature journals. The challenge was to obtain sufficient soluble, native-like material for structural studies. Over a 12 year collaboration with John Moore’s group at Weill Medical College, we were able to determine the crystal structure of a soluble cleaved Env trimer (SOSIP) (1) that enabled a view of the interaction between the component gp41 and gp120 subunits, assembly of the three gp140s that compose the trimer, and its interaction with bnAbs. This work with a companion cryo-EM structure of the same trimer (Lyumkis et al., Science 342:1484-90, 2013) defined key features of the Env trimer and that it was a faithful mimic of Env on the virus, and both papers were cited as runner up papers of the year in Science in 2014. Our current focus is how HIV-1 Env is recognized by bnAbs to define sites of vulnerability for structure-assisted vaccine design (e.g. Pejchal et al., Science 334:1097-1103, 2011 and 2). These bnAbs almost all also target glycans on the Env surface (3). The Env structure has propelled its use as a candidate immunogen and many groups are using this soluble BG505 SOSIP Env trimer construct for vaccine design, such as NFL here, and for testing in animal models. We generated responses against the SOSIP trimer as a candidate immunogen in small animal models and, for the first time, a tier-2 neutralizing response against the homologous virus (Sanders et al., Science 349:aac4223, 2015), thus showing progress towards design of an HIV-1 vaccine (reviewed 4). Indeed, this BG505 SOSIP Env trimer has entered a Phase 1 human clinical trial (March 2019).
Select Publications
- Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 2013; 342:1477-83. PMCID: PMC3886632.
- Structural evolution of glycan recognition by a family of potent HIV antibodies. Cell 2014; 159:69-79. PMCID: PMC4278586.
- Structure and immune recognition of the HIV glycan shield. Annu Rev Biophys. 2018 Mar 29. doi: 10.1146/annurev-biophys-060414-034156. PMCID: PMC6163090.
- The HIV-1 Envelope glycoprotein structure: nailing down a moving target. Immunol. Rev. 2017; 275:21-32. PMCID: PMC5300090.
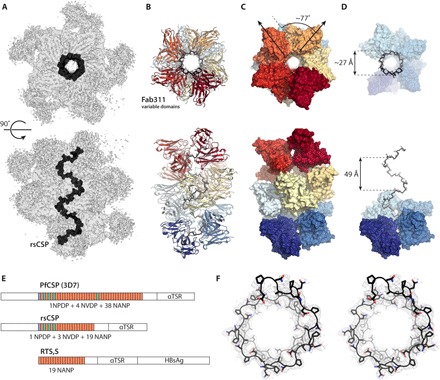 Malaria P. falciparum CSP structure, protective antibodies, sites of vulnerability and structure-assisted vaccine design. We have determined over 20 structures of protective antibodies to the CSP of falciparum by x-ray crystallography and through collaborations with Prof. Andrew Ward by negative stain- and cryo-electron microscopy. We have identified the binding epitopes (that include the NANP repeats, junctional epitope, and C-terminal domain), common binding motifs, and preferred binding interactions of protective antibodies through structural and other biophysical analyses. We have identified the structural basis of restricted germline usage for protective antibodies to the NANP repeats. We have also shown how evolution of unusual homotypic inter-Fab contacts in some of these antibodies helps provide avidity to the interactions and allows for the very close proximity of the antibodies that decorate the long-range spiral structure of the NANP repeats in CSP. This information is now being used for vaccine design.
Malaria P. falciparum CSP structure, protective antibodies, sites of vulnerability and structure-assisted vaccine design. We have determined over 20 structures of protective antibodies to the CSP of falciparum by x-ray crystallography and through collaborations with Prof. Andrew Ward by negative stain- and cryo-electron microscopy. We have identified the binding epitopes (that include the NANP repeats, junctional epitope, and C-terminal domain), common binding motifs, and preferred binding interactions of protective antibodies through structural and other biophysical analyses. We have identified the structural basis of restricted germline usage for protective antibodies to the NANP repeats. We have also shown how evolution of unusual homotypic inter-Fab contacts in some of these antibodies helps provide avidity to the interactions and allows for the very close proximity of the antibodies that decorate the long-range spiral structure of the NANP repeats in CSP. This information is now being used for vaccine design.
Select Publications
- Structural basis for antibody recognition of the NANP repeats in Plasmodium falciparum circumsporozoite protein. Proc Natl Acad Sci U S A 2017;114:E10438-E10445. PMCID: PMC5715787.
- Cryo-EM structure of falciparumcircumsporozoite protein with a vaccine-elicited antibody is stabilized by somatically mutated inter-Fab contacts. Sci Adv 2018;4:8529. PMCID: PMC6179375.
- Diverse antibody responses to conserved structural motifs in Plasmodium falciparum circumsporozoite protein. J Mol Bio 2019;19:30738-7. PMID: 31883801.
- Structure and mechanism of monoclonal antibody binding to the junctional epitope of Plasmodium falciparumcircumsporozoite protein. PLoS Pathog 2020;16:E1008373. PMID: 32150583.
HCV antigen structure and complexes with broadly neutralizing antibodies. We determined the first crystal structure of the E2 glycoprotein core from HCV in complex with bnAb AR3C in collaboration with Dr. Mansun Law and negative-stain EM structures of full-length E2 with the CD81 receptor with Dr. Andrew Ward (a). These studies revealed an unexpected compact architecture of the E2 protein and identified the receptor binding site and sites of potential vulnerability on the virus. We have also determined structures for bnAbs in complex with linear HCV E2 peptides corresponding to a highly conserved antigenic sites (Kong et al. 2012, and b). We also have determined other crystal structures of E2 and bnAbs to E2 as well as functional and genetic insights (c). These insights have provided valuable information for structure-assisted vaccine design (d).
Select Publications
- Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013;342:1090-4. PMCID: PMC3954638.
- Immunogenetic and structural analysis of a class of HCV broadly neutralizing antibodies and their precursors. Proc Natl Acad Sci USA. 2018;115:7569-74. PMCID: PMC6055136.
- Genetic and structural insights into broad neutralization of hepatitis C virus by human VH1-69 antibodies. Sci Adv. 2019;5:eaav1882. PMCID: PMC6314831.
- Capitalizing on knowledge of hepatitis C virus neutralizing epitopes for rational vaccine design. Curr Opin Virol 2015;11:148-57. PMCID: PMC4507806.
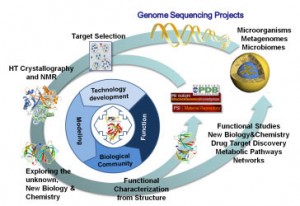 Exploring the Expanding Protein Universe. The Joint Center for Structural Genomics (JCSG) was a multi-institutional consortium based at TSRI, with major components the Genomics Institute of the Novartis Research Foundation, the University of California San Diego, the Sanford-Burnham Medical Research Institute, and the Stanford Synchrotron Radiation Lightsource (SSRL) at Stanford University. The JCSG pioneered new and improved HTP gene to structure methods and technologies and designed tools to predict the probability of protein crystallization from the primary sequence (1) that have increased crystallization success. Our pipeline was highly efficient and scalable and very productive (2) and successfully overcame major challenges such as determining the complete central metabolic network for an organism (3), exploring new regions of the protein universe (4) and providing novel insights into domains of unknown function. We have also collaborated with many investigators to solve structures of great interest to them. We continue to use the equipment, technologies and methodologies that we developed in the JCSG, which are now operated by the Wilson lab.
Exploring the Expanding Protein Universe. The Joint Center for Structural Genomics (JCSG) was a multi-institutional consortium based at TSRI, with major components the Genomics Institute of the Novartis Research Foundation, the University of California San Diego, the Sanford-Burnham Medical Research Institute, and the Stanford Synchrotron Radiation Lightsource (SSRL) at Stanford University. The JCSG pioneered new and improved HTP gene to structure methods and technologies and designed tools to predict the probability of protein crystallization from the primary sequence (1) that have increased crystallization success. Our pipeline was highly efficient and scalable and very productive (2) and successfully overcame major challenges such as determining the complete central metabolic network for an organism (3), exploring new regions of the protein universe (4) and providing novel insights into domains of unknown function. We have also collaborated with many investigators to solve structures of great interest to them. We continue to use the equipment, technologies and methodologies that we developed in the JCSG, which are now operated by the Wilson lab.
Select Publications
- XtalPred: a web server for prediction of protein crystallizability. Bioinformatics 2007; 23:3403-5. PMID: 17921170.
- The JCSG high-throughput structural biology pipeline. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010; 66:1137-42. PMCID: PMC2954196.
- Three-dimensional structural view of the central metabolic network of Thermotoga maritima. Science 2009; 325:1544-9. PMCID: PMC2833182.
- Exploration of uncharted regions of the protein universe. PLoS Biol. 2009 Sep;7(9):e1000205. PMCID: PMC2744874.
Mission of the JCSG
The JCSG high-throughput (HTP) platform, assembled over 15 years, delivered a large numbers of protein structures to the community by both X-ray crystallography and NMR on a wide range of targets from bacteria to human, including challenging targets, such as eukaryotic proteins, protein-protein and other macromolecular complexes. Our primary approach to HTP structural biology involves processing sets of targets through an extensive combination of bioinformatics and biophysical analyses to efficiently characterize each target in order to optimize its path through our pipeline. During PSI:Biology, the 3nd phase of the NIGMS Protein Structure Initiative, the JCSG leveraged its HTP platform to promote the biological and biomedical impact of all of our structures through extensive functional and bioinformatics analyses both internally and in collaboration with our Partnerships, community nominated targets and via other collaborations within the PSI:Biology network, as well as the general scientific community. Using a more focused approach, in close collaboration with our assigned Partnership Centers, we tackled much more challenging targets, such as protein-protein and protein-nucleic acid complexes and capitalizing on our extensive experience to develop the best strategies to enhance chances of success. In parallel, we continued to process our internal biomedical and structure coverage targets in a HTP mode. These target sets were focused on the microbial communities that inhabit specific niches and environments of the human body. Interactions of commensal bacteria with the human body are profound and have a significant impact on maintenance of general human health, as well as being associated with disease. The human microbiota are dominated by poorly characterized bacterial phyla, which contain an unusually high number of uncharacterized proteins that so far remain largely unstudied. Thus, this biological theme project served a dual purpose in also addressing coverage of novel sequence space as a component of the central PSI structural coverage theme. The influence of the microbiome upon human development, physiology, immunity, and nutrition is only starting to surface and, thus, represented an exciting new frontier for high throughput structural biology where investigated the contributions of these microorganisms to human health, as well as to disease. We are mainly focused on the human gut microbiome. As part of the PSI:Biology Network, we strived to promote widespread use of PSI resources, materials, methodologies and data to the general scientific community. The JCSG also continued to develop new technologies and methodologies, both experimental and computational, to address a wide spectrum of targets, while keeping the cost per structure to a minimum and quality to the highest standard. We also continued to contribute to the original PSI mission of structural coverage of the expanding protein universe.